合成生物學作為迅速發展的交叉學科,已在生物醫藥、新材料、診斷、能源和數據儲存等領域展現越來越廣泛的應用潛力。合成基因組學作為合成生物學的重要研究方向,著重于新生命體系的從頭設計與合成,其以Oligo(寡核苷酸鏈)合成、拼裝和轉移等核心技術為支撐。新生命體系的從頭設計與合成不僅需要合成組成基因組的小片段DNA片段,還需要通過后續的組裝與拼接獲取完整的合成基因組。Oligo合成及基因組DNA拼裝、轉移技術是合成基因組學乃至整個合成生物學領域的核心技術體系,今天主要來介紹體外單鏈DNA(Oligo)合成與雙鏈DNA拼接組裝技術。
1. 單鏈Oligo合成技術 Oligo即寡核苷酸鏈,分子實驗室常用的PCR引物、NGS捕獲探針、qPCR探針和FISH探針等都是Oligo。與雙鏈DNA相比,Oligo有個一個共同的特點他們都是單鏈DNA序列,根據功能不同,可以在Oligo上修飾熒光基團、化學基團(如,羧基、氨基、巰基等,)等。根據合成原理,目前Oligo的合成方法可分為已經成熟并商業化的化學合成法和正在研發中的酶促合成法。
1.1 化學法 Oligo的化學合成研究始于20世紀50年代,Michelson和Todd首次報道采用磷酸二酯法實現了寡聚二核苷酸的合成。到20世紀80年代,Beaucage和
Caruthers開發了基于亞磷酰胺的DNA合成方,即今天Oligo自動化生產所采用的主要方法。該方法包括去保護、偶聯、加帽(可選)及氧化 4 個步驟 (圖 1)。由于隨著鏈延長所帶來的化學反應效率、合成純度以及產率的下降,目前該方法合成的Oligo長度一般不會超過200個核苷酸(nt)。為了提高通量,從20世
紀90年代開始發展起來的基于微陣列芯片的DNA合成策略,使得合成成本降低了若干個數量級。然而由于芯片特有的不均一性及邊緣效應等原因,相較傳統的柱法合成,芯片法合成在長度、準確性方面均有所下降。為了增加芯片合成的精確度,科學家采用不同的技術策略,從各異的芯片產物中挑選出正確合成的寡核苷酸原料。Kosuri采用了多重PCR策略,選擇性擴增目的寡核苷酸片段,結合酶促糾錯等方法,可以合成長度超過200
nt的寡核苷酸原料,實現了更大規模和更高精度的合成。 圖 1.固相亞磷酰胺法從頭合成寡聚核苷酸鏈的4步反應 4步反應包括:(1)去保護。酸催化去除
DMT(二甲氧基三苯基甲基)基團,以便下一輪堿基(dA、dC、dG和dT)添加。(2)堿基偶聯。將含有DMT保護基團的亞磷酰胺通過四唑活化劑加到未保護的5′OH末端。(3)加帽。將游離的5′OH乙酰化,以防止進一步的鏈延伸所造成的單堿基缺失。(4)氧化。通過碘液將磷酸三酯氧化為磷酸鹽,進入一個反應循環.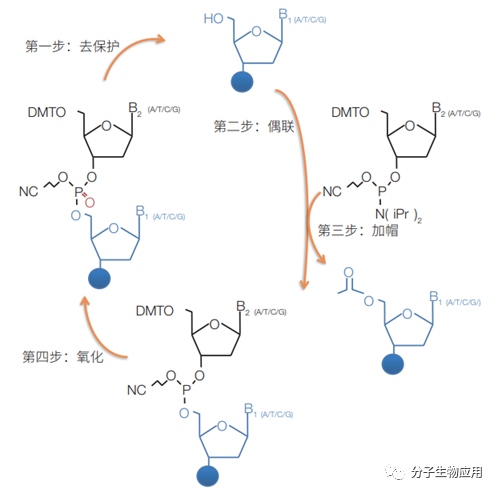
1.2 酶促從頭合成法 目前亞磷酰胺法是商業化Oligo合成的主流方法,但其合成的長度限制在200nt左右,而且合成過程中亦會大量使用有毒化學試劑。由于在合成準確性及不需要使用毒性化合物等方面的潛在優勢,酶促合成方法受到了越來越多的關注。與化學法相比,酶促法的作用條件溫和,對DNA的損傷較少,有助于準確性的提高,同時減少了副產物的產生,可實現更長Oligo的合成。然而,酶促法的發展緩慢,至今未能實現商業化。根據Jensen和Davis對DNA酶促從頭合成發展的總結,末端脫氧核苷酰轉移酶(TdT)介導的酶促合成法是較好的選擇,但還需要更進一步優化。TdT介導的DNA合成技術開發中首先需要解決單個dNTP添加后的終止效率及末端重新活化的問題。近期,Palluk等提出了一種解決方案,他們將
dNTP通過可光誘導剪切的連接子與TdT連接,所得dNTP-TdT復合物可在10—20s的時間完成DNA鏈的延伸,而且可以重復進行,實現特定DNA鏈的合成。該方法將為具有實際應用價值的酶促DNA合成法的開發打下基礎。
2. 雙鏈DNA/基因合成技術 DNA合成技術是合成基因組學,乃至現代分子生物學的根基,受當前合成技術的限制,雙鏈DNA/目標基因組只能以短鏈Oligo的形式逐步拼接才能獲得。PCR(聚合酶鏈式反應)或者酶切方法只限于獲得自然界已有的DNA片段,而雙鏈DNA/基因組的從頭合成則需要通過Oligo的拼接獲得,所以,如何提高Oligo合成的長度和效率、降低合成成本是后續進行大規模基因組合成的關鍵突破點。體外雙鏈DNA的拼接根據原理區別,分為以下幾種技術。
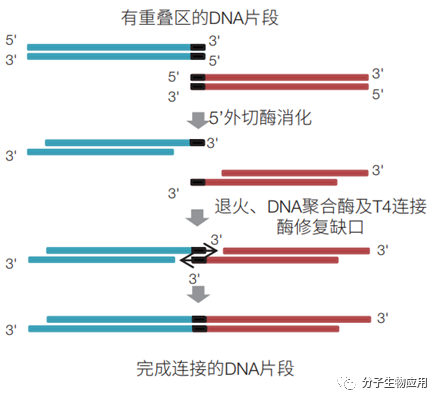
2.1 Gibson 組裝 Gibson組裝最早由Gibson等在2009年提出,原理如圖2所示。該技術基于5′核酸外切酶、DNA 聚合酶及連接酶,利用DNA片段的兩端同源序列(其長度通常為15?40bp),實現DNA組裝。將這些DNA片段和3種酶的混合溶液孵育1h即可。5’外切酶,從5’端開始對DNA進行消化,產生長的黏性末端,這樣便于與另外的同源末端進行配對結合;DNA聚合酶,用于修補由5’外切酶產生的缺口,在Gibson組裝中用的是Phusion
DNA聚合酶,當然,Taq
DNA聚合酶也可以用;DNA連接酶,實現共價連接,形成完整的DNA分子。該方法的優勢在于這3種酶都可以在同一個溫度下發揮功能,可一步完成組裝,組裝后的質粒可直接用于轉化感受態細胞,無需限制性內切酶,通常該方法可組裝不超過6個片段。 2010
年,Gibson等科學家使用Gibson組裝和釀酒酵母體內同源重組人工合成了1.08Mb的絲狀支原體基因組并轉入去核的山羊支原體,新移殖的細胞表現出了絲狀支原體的性狀。2016年,Hutchison
等人先用 PCR 合成一系列絲狀支原體基因組片段,每個DNA片段長1.4 kb,然后每5個分成一組,再利用 Gibson
組裝技術將5個一組的DNA片段連接成7 kb的單片段,最后組裝成完整的基因組。作者只保留了必需基因,將1.08
Mb的基因組縮短到了含有473個基因的 531 kb。2017 年,Esvelt等人將Gibson組裝技術與CRISPR/Cas9
技術結合構建了一個PAM質粒文庫,用于評估鑒定來自不同物種的一系列不同的Cas9蛋白的活性。該方法在體外組裝技術中應用廣泛。 圖2 基于5′核酸外切酶、DNA 聚合酶及連接酶的Gibson組裝法 通過一步等溫體外重組兩個相鄰的DNA片段(紅色和藍色,共用終端序列為黑色),兩個DNA片段共價連接成同一個DNA分子。T5外切酶去除雙鏈DNA分子的5’端核苷酸,產生粘性末端,互補單鏈DNA退火結合在一起,Phusion
DNA聚合酶填充缺口,Taq DNA連接酶密封缺口。T5外切酶是不耐熱的,在50℃培養期間失活,所以不會與DNA聚合酶競爭DNA模板。